1. Khái niệm
Hội chứng XYY hay còn gọi là một trong các hội chứng sau:
- Hội chứng 47,XYY
- Hội chứng Jacob (Jacob’s syndrome)
- Hội chứng siêu nam (supermale)
- Hội chứng YY
Hội chứng XYY là một trong các bệnh rối loạn nhiễm sắc giới tính hiếm gặp ở người nam, bộ nhiễm sắc thể giới tính ở người nam có thêm 1 nhiễm sắc Y. Người nam bình thường chỉ có 1 nhiễm sắc thể X và 1 nhiễm sắc thể Y, người bệnh có 1 nhiễm sắc thể X và 2 nhiễm sắc thể Y. Người bệnh thường cao, giai đoạn thiếu niên, mụn trứng cá mọc nhiều đến nghiêm trọng, người bệnh có biểu hiện chậm phát triển tâm thần, khuyết tật về học tập, ngôn ngữ và hành vi, hay bốc đồng, trí tuệ ở mức bình thường hoặc thấp hơn 10 - 15 điểm.
Công thức nhiễm sắc thể (Karyotype) của người người bệnh là 47,XYY.
Tỷ lệ mắc hội chứng XYY ở trẻ nam mới sinh là 1:750 - 1:1.000.
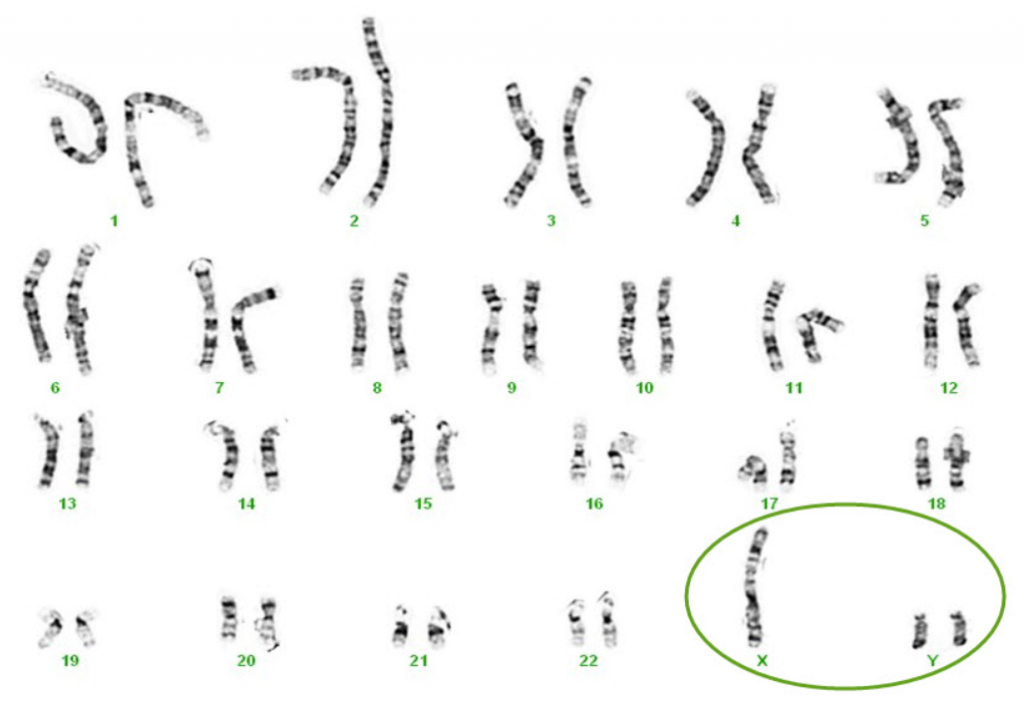 Karyotype của người bệnh nam mắc hội chứng Jacob (47,XYY)
Karyotype của người bệnh nam mắc hội chứng Jacob (47,XYY)
2. Nguyên nhân, cơ chế bệnh sinh
Nam giới bị mắc hội chứng XYY là do sự rối loạn phân ly của nhiễm sắc thể giới tính của người bố trong giảm phân tạo tinh trùng YY thay vì tinh trùng Y. Tinh trùng YY kết hợp với trứng X của mẹ, tạo hợp tử XYY phát triển thành cơ thể mắc hội chứng XYY. Ngoài ra, một số ít trường hợp, sự rối loạn phân li xảy ra ở hợp tử hay phôi bào dẫn đến cơ thể bị khảm có đồng thời hai dòng tế bào 46,XY và 47,XYY. Khi đó công thức nhiễm sắc thể của người mắc hội chứng XYY là 46,XY/47,XYY.
3. Chẩn đoán
a. Triệu chứng lâm sàng
Nam giới mắc hội chứng XYY thường bị bỏ qua chẩn đoán, phần lớn khi lập gia đình có vấn đề về chức năng sinh sản như hiếm muộn, người bệnh mới được phát hiện mình mắc bệnh. Một số đặc điểm lâm sàng lưu ý:
- Thể trạng cao, tăng nhanh và rõ khi trẻ 5 - 6 tuổi.
- Nhiều mụn trứng cá
- Có thể có khả năng sinh sản hoặc tình dục bình thường
- Trẻ nhỏ mắc hội chứng XYY có thể chậm phát triển tâm thần nhẹ, khuyết tật về học tập chiếm tới 50% các trường hợp, phần lớn là chậm nói và các vấn đề về ngôn ngữ, khó phát âm.
- Đặc điểm nổi trội của của người nam mắc hội chứng XYY liên quan tới hành vi như tính bùng nổ, hiếu động, bốc đồng, hành động thách thức hoặc trong một số trường hợp là hành vi chống đối xã hội, còn được cho là kiểu hình tội phạm. Người bệnh có thể biểu hiện giảm chú ý, tăng động và nguy cơ mắc chứng rối loạn phổ tự kỷ.
 Triệu chứng lâm sàng của hội chứng Jacob (47,XYY)
Triệu chứng lâm sàng của hội chứng Jacob (47,XYY)
b. Cận lâm sàng
- Karyotype: thể thuần 47,XYY hoặc thể khảm 46,XY/47,XYY.
- Xét nghiệm tinh dịch đồ: ít tinh trùng (thiểu tinh) hoặc có thể tinh dịch đồ bình thường.
c. Chẩn đoán xác định
Người nam có karyotype là 47,XYY ở dạng thuần hoặc thể khảm.
d. Chẩn đoán phân biệt
- Hội chứng Klinefelter, karyotype là 47,XXY (thừa 1 nhiễm sắc thể giới tính X).
- Hội chứng Sotos: đây là một rối loạn di truyền đặc trưng bởi sự tăng trưởng quá mức trước và sau khi sinh. Một trong những đặc điểm chính của hội chứng Sotos là biểu hiện đặc biệt trên khuôn mặt bao gồm đỏ bừng mặt, trán dô, mắt xếch, hàm hẹp, khuôn mặt hẹp dài và hình dạng đầu tương tự như một quả lê ngược. Chiều cao và chu vi đầu lớn hơn trung bình đối với hầu hết trẻ em bị ảnh hưởng. Người bệnh bị chậm phát triển vận động và ngôn ngữ cũng như chậm phát triển tâm thần từ nhẹ đến nặng. Các vấn đề khác liên quan đến hội chứng Sotos bao gồm vàng da ở trẻ sơ sinh, cột sống cong (vẹo cột sống), co giật, lác mắt (strabismus), mất thính lực dẫn truyền, dị tật tim bẩm sinh, bất thường về thận và các vấn đề về hành vi. Nguyên nhân gây hội chứng Sotos do đột biến gen NSD1.
- Hội chứng Marfan ở người nam, biểu hiện người cao, chân tay dài. Đây là rối loạn di truyền do đột biến gen FBN1 quy định tổng hợp fibrillin-1 liên quan chặt chẽ tới mô liên kết. Tim mạch và hệ thống cơ xương khớp bị ảnh hưởng nhiều nhất. Các triệu chứng chính bao gồm sự phát triển quá mức của xương cánh tay và xương đùi, cong vẹo cột sống, thành ngực biến dạng (pectus), trật khớp ống kính của mắt (lạc vị thể thủy tinh - lectis lentis), cận thị, phình động mạch và bóc tách động mạch chủ, hở van 3 lá và van động mạch chủ, …
4. Điều trị
- Việc sửa chữa dư thừa 1 nhiễm sắc thể Y là không thực hiện được. Điều trị triệu chứng giúp cho người bệnh hòa nhập với cuộc sống.
- Tư vấn di truyền và liệu pháp tâm lý cho người bệnh và người nhà người bệnh, giúp họ hòa nhập với cộng đồng.
Xem thêm: Hội chứng Klinefelter